
洁净室标准在医疗器械制造中起着至关重要的作用。这些受控环境可帮助您保持无菌并防止污染。当您遵循这些标准时,您可以确保医疗器械满足严格的安全和质量要求。例如,洁净室注塑成型使您能够制造出具有最小杂质风险的精密组件。通过维护洁净室方案,您可以提高产品可靠性并保护患者健康。
洁净室标准对于维持污染风险降至最低的受控环境至关重要。这些标准定义了空气清洁度、颗粒物控制和环境监测的协议和要求。通过遵守洁净室标准,您可以确保在无菌和受控的环境中制造医疗器械。这降低了污染风险,提高了产品的整体质量。
洁净室旨在控制空气中的颗粒、温度、湿度和压力。例如,HEPA 过滤器可去除 99.97% 的小至 0.3 微米的颗粒,而 ULPA 过滤器可实现更高的效率,可捕获 99.999% 的 100 纳米以下颗粒。这些过滤系统在保持环境清洁方面发挥着关键作用。
两项关键法规指导医疗器械生产中的洁净室实践:ISO 14644 和 ISO 13485。ISO 14644 侧重于空气清洁度,并为控制洁净室中的空气传播颗粒提供了框架。它建立了监测和维持生物污染物水平的方法。例如,ISO 14644 根据每立方米的颗粒浓度对洁净室进行分类,范围从 ISO 1 级(最清洁)到 ISO 9 级。
另一方面,ISO 13485 强调医疗器械的质量管理体系。它确保您的制造过程符合法规要求,并生产出安全、高质量的产品。通过遵循 ISO 13485,您可以证明符合全球标准,并与客户和监管机构建立信任。
| 标准 | 重点领域 |
|---|---|
| ISO 14644 认证 | 生物污染物去除方法 |
| ISO 13485 认证 | 医疗器械质量管理 |
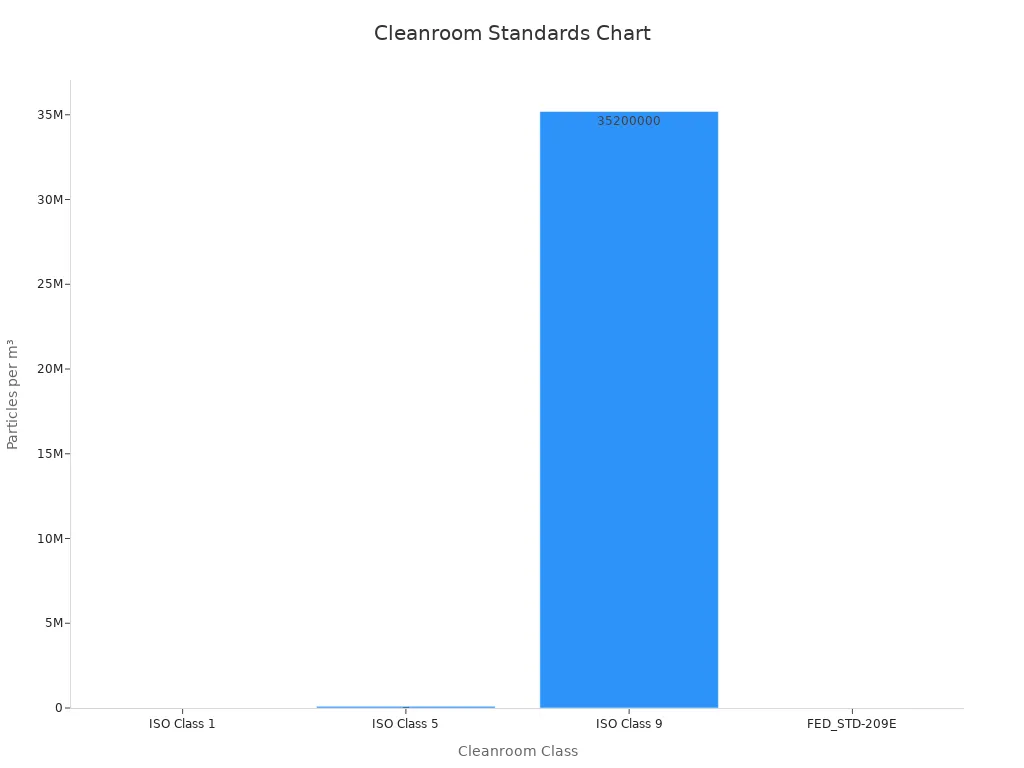
洁净室分类基于特定尺寸范围内空气中颗粒的浓度。这些分类可帮助您确定制造流程所需的清洁度级别。例如,ISO 1 级洁净室每立方米只允许 10 个大于 0.1 微米的颗粒,而 ISO 9 级洁净室与普通室内空气相当。大多数医疗器械制造洁净室都属于 ISO 7 级或 ISO 8 级。
| 洁净室类 | 粒径 (μm) | 每立方米的颗粒数 |
|---|---|---|
| ISO 1 级 | 0.1 | 10 |
| ISO 5 级 | 0.1 | 100,000 |
| ISO 9 级 | 0.5 | 35,200,000 |
保持空气质量控制对于满足这些分类至关重要。先进的过滤系统,如 HEPA 和 ULPA 过滤器,确保洁净室符合要求的标准。例如,7 级洁净室的最大颗粒浓度为每立方米 352,000 个颗粒。定期监测和验证空气质量有助于您保持对洁净室分类的合规性,并确保制造过程的完整性。

防护服和卫生程序对于保持洁净室的清洁至关重要。您必须遵循严格的协议,以防止洁净室注塑成型过程中的污染。正确的礼服包括穿着专门的服装,例如工作服、手套、口罩和鞋套,旨在尽量减少体内颗粒的释放。这些服装充当屏障,确保皮肤薄片、头发和灰尘等污染物不会进入洁净室环境。
进入洁净室前,应对双手进行彻底清洁和消毒。此步骤降低了将微生物或颗粒引入受控环境的风险。礼服必须在指定区域进行,通常称为礼服室,您可以在那里按特定顺序穿上洁净室认可的服装。例如,您可以从鞋套开始,然后是手套,然后是全身套装。该顺序可确保最大程度的卫生并最大限度地降低污染风险。
卫生规程不仅限于穿礼服。定期清洁和消毒表面、工具和设备对于保持无菌环境是必要的。您还应该遵守严格的行为准则,例如避免不必要的动作和轻声说话,以减少颗粒的产生。这些做法确保洁净室始终符合清洁标准,从而提高所生产医疗器械的质量。
正确的材料处理和储存对于保持洁净室注塑成型所用材料的完整性至关重要。您必须确保所有进入洁净室的材料都经过彻底清洁和灭菌。此步骤可防止引入可能影响制造过程的污染物。过滤器和疏水阀通常用于消除物料输送过程中的空气颗粒,确保安全清洁的生产环境。
进入洁净室后,材料应存放在符合清洁度标准的指定区域。例如,密封容器或橱柜可以保护材料免受空气中颗粒的影响。您还应该标记和整理材料,以促进可追溯性并防止交叉污染。定期检查存储区域有助于保持对洁净室规程的遵守。
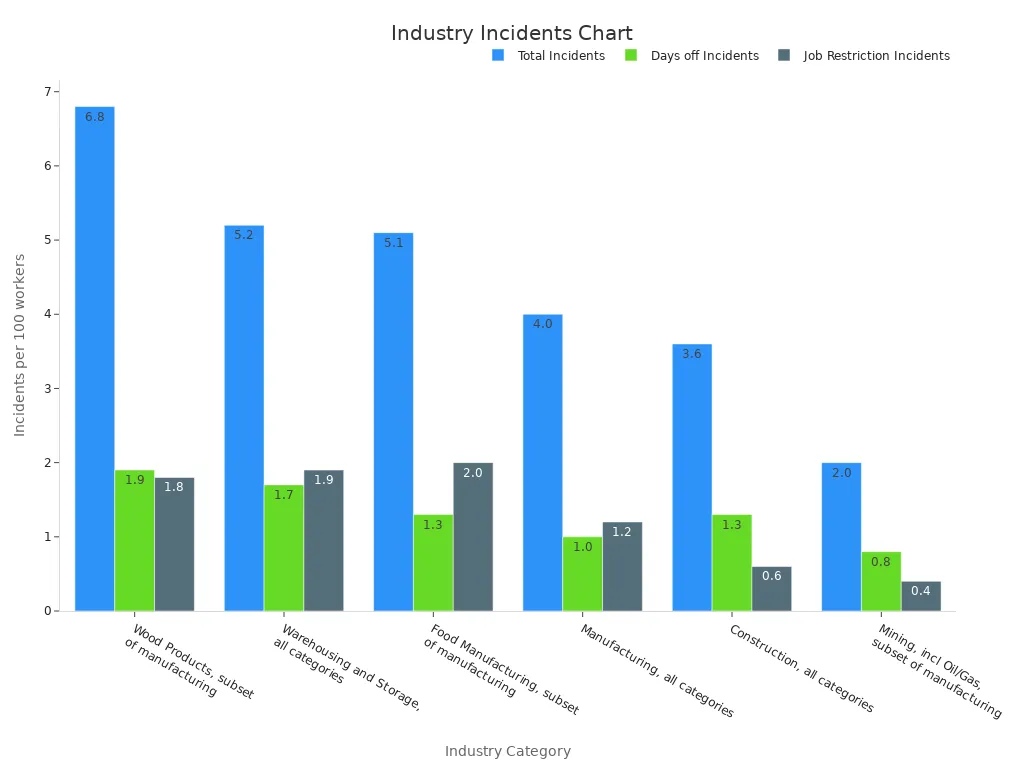
统计数据强调了高效物料处理和储存程序的重要性。例如,与其他行业相比,具有严格处理协议的行业(如制造和仓储)报告的事故率较低。
| 行业类别 | 每 100 名工人的事故总数 | 每 100 名工人中无休息日的事故数 | 每 100 名工人中具有工作调动或限制的事件数 |
|---|---|---|---|
| 制造,所有类别 | 4.0 | 1.0 | 1.2 |
| 仓储 - 所有类别 | 5.2 | 1.7 | 1.9 |
| 食品制造,制造的子集 | 5.1 | 1.3 | 2.0 |
通过实施强大的材料处理和储存程序,您可以降低污染风险、提高效率并确保医疗器械的质量。
设备维护和验证对于确保洁净室运行的可靠性至关重要。您必须定期检查和维护设备,以防止可能影响清洁度的故障。验证过程确认设备和系统符合要求的规格,并在正常作条件下按预期运行。
验证过程通常包括几个步骤:
-设计资格 (DQ):确认洁净室设计符合行业标准。
-安装资格 (IQ):验证设备是否安装正确。
-作资格 (OQ):在正常条件下测试设备以确保正常功能。
-性能鉴定 (PQ):评估长期绩效以维持清洁标准。
持续的环境监测是验证的另一个关键方面。您应该定期检查空气质量、温度、湿度和颗粒物水平,以确保符合洁净室标准。微生物测试还可以识别潜在的污染源,使您能够及时采取纠正措施。
维护和验证活动的全面文档对于法规遵从性至关重要。详细的记录表明您对质量的承诺,并提供遵守行业标准的证据。通过优先考虑设备维护和验证,您可以提高洁净室作的可靠性并生产高质量的医疗设备。
可追溯性是医疗器械制造质量控制过程的基石。它允许您跟踪生产中使用的每个组件和材料,确保符合监管标准。通过实施强大的可追溯性系统,您可以在潜在问题升级之前识别并解决它们。例如,洁净室环境中的环境监测有助于检测污染源,例如空气或表面颗粒,并确保产品安全。
一项案例研究显示,对洁净室环境(包括空气、表面和压缩空气等公用设施)进行全面采样,显著提高了可追溯性。这种方法减少了实验室错误并最大限度地减少了制造故障。
可追溯性还支持验证和审计流程。生产中涉及的材料、设备和人员的详细记录提供了清晰的审计跟踪。这种透明度不仅提高了合规性,还建立了与监管机构和客户的信任。
在洁净室环境中保持最佳条件对于确保产品质量和合规性至关重要。您可以通过监控关键指标(例如空气传播的颗粒、温度、湿度和微生物污染)来实现这一点。
| 测量指标 | 描述 |
|---|---|
| 空气颗粒物监测 | 对空气中的颗粒进行连续采样和定量,以保持清洁度标准。 |
| 环境参数监测 | 监测温度、湿度和压力,以确保最佳的洁净室条件。 |
| 微生物监测 | 通过自动采样方法检测和分析微生物污染风险。 |
| 防护服室监控 | 确保遵守防护服规程,以防止人员污染。 |
定期监测可帮助您识别可能损害洁净室完整性的趋势和偏差。例如,统计过程控制 (SPC) 方法(如控制图)允许您跟踪变异性并保持一致的质量。通过及时解决问题,您可以防止代价高昂的生产延迟,并确保符合监管标准。
准确的文档记录对于满足监管合规要求至关重要。它在审计和检查期间用作证据,保护您的运营免受罚款和不合规风险。全面的记录表明您对质量的承诺和对行业标准的遵守。
主要文档类型包括:
- 清理日志,详细说明使用的清理活动、人员和代理。
- 设备校准记录,以确保工具正常运行。
- 记录整个制造过程以实现可追溯性的批次记录。
此外,维护设计文档、资格协议和环境监测日志可以加强您的合规工作。这些记录支持验证和审计流程,确保您的洁净室运营符合监管期望。通过优先考虑文档和可追溯性,您可以增强产品安全性并与利益相关者建立信任。

洁净室对于确保产品安全和质量是必不可少的。它们显著降低了污染风险,使其成为医疗器械制造的基石。
提示:遵守洁净室实践不仅可以提高效率,还可以与客户和监管机构建立信任。当您优先考虑洁净室方案时,您就为医疗器械制造的成功奠定了基础。这些做法可帮助您实现合规性、提高产品质量并保护患者健康。
提示:定期监控还可以及早发现潜在问题,从长远来看可以节省时间和成本。

RM1223, 12F, No.1, Fuji Bldg, No. 6018 Longgang RD. , Longgang Dist., Shenzhen,China.


Tel: 0086-755-89618186

Contact: Paul Hu 0086-18675501028







 LEAVE MESSAGE
LEAVE MESSAGE